El 16 de diciembre de 2025 la Comisión Europea publicó su propuesta para revisar el Reglamento de Productos Sanitarios (MDR) y el Reglamento de Diagnóstico In Vitro (IVDR), ya bautizados en el sector como MDR 2.0 e IVDR 2.0. El paquete no llega solo: también “toca” la Ley de IA (AI Act) para ordenar solapamientos y aclarar interacciones con otras normas.
La Comisión, por fin, pone negro sobre blanco algo que el sector lleva años describiendo con menos diplomacia: el sistema MDR/IVDR funciona, pero funciona con fricción. Falla en lo que más condiciona a un equipo de desarrollo: coste total, plazos reales y predictibilidad. La combinación de procedimientos exigentes, capacidad limitada de evaluación (organismos notificados y autoridades), y una coordinación que no siempre es homogénea entre Estados miembros y entre evaluadores, ha generado un fenómeno que ya no se puede tratar como anecdótico: productos que se retrasan, se rediseñan para “encajar” en una clasificación de riesgo más liviana o directamente se retiran antes de llegar al mercado europeo. Y esto no ocurre solo con tecnologías radicales; afecta de forma desproporcionada a tres categorías especialmente sensibles: nichos clínicos, dispositivos huérfanos y mejoras en los dispositivos que aportan valor pero no justifican, desde una lógica empresarial, un ciclo regulatorio largo, caro e incierto.
Desde la perspectiva de I+D, el problema no es únicamente “más requisitos”. Es el riesgo de variabilidad: que el esfuerzo invertido en diseño, evidencia y dossier no se traduzca en un resultado evaluador razonablemente predecible. Cuando la ruta regulatoria se percibe como una cadena de decisiones poco trazables (o demasiado dependientes de interpretaciones), el efecto en el producto es inmediato. El diseño se vuelve conservador: se evita lo novedoso, se limitan claims, se eligen arquitecturas técnicas menos ambiciosas, y se minimizan cambios que puedan disparar preguntas de equivalencia, clasificación o evidencia clínica. Además, se desplazan recursos a tareas defensivas (documentación, re-trabajo, estudios “por si acaso”) y se produce un retraso casi estructural en la transferencia a fabricación, porque nadie quiere congelar diseño y escalar producción con incertidumbre sobre el “qué más” pedirá la evaluación.
En ese contexto, MDR/IVDR 2.0 se plantea como un intento de reducir incertidumbre sin desregular: simplificar donde la complejidad no añade seguridad proporcional, y crear mecanismos más adaptativos para no castigar a quien desarrolla para poblaciones pequeñas o necesidades no cubiertas. Pero aquí está la tensión: mientras se habla de simplificación y proporcionalidad, la propuesta también refuerza el uso de herramientas de la Comisión (actos delegados y de ejecución) para ajustar requisitos ante progreso científico, desarrollos internacionales o tecnologías emergentes (con la IA como caso paradigmático). Para el fabricante, esto tiene una lectura doble. Por un lado, puede aportar agilidad: adaptar el marco sin esperar años a una reforma completa. Por otro, introduce un elemento de dinámica regulatoria que afecta directamente al diseño: si el “estado del arte” y ciertos requisitos pueden ser modificados por actos posteriores, la estrategia de desarrollo debe ser más robusta, más modular y con mayor capacidad de absorción de cambios, especialmente en software, ciberseguridad y sistemas con IA.
Cambios que afectan al “diseño regulatorio” del producto
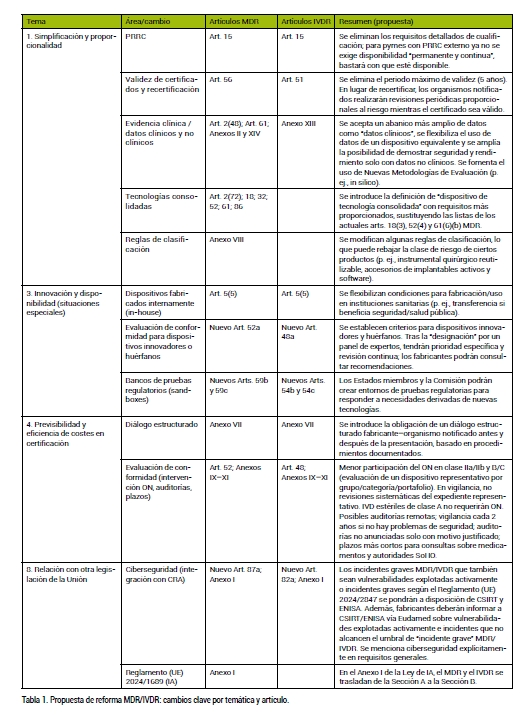
La tabla 1 recoge los aspectos más relevantes de la propuesta de reforma. A continuación, vamos a proceder a desarrollar algunos de ellos.
Equivalencia: de “idéntico” a “similar”
Una de las palancas más potentes para acelerar el desarrollo de nuevos productos es la equivalencia. Hasta ahora, demostrar la equivalencia se había convertido en una quimera: requisitos de acceso a documentación del “equivalente”, exigencias de similitud casi milimétrica en la arquitectura del producto, y la causa de debates interminables sobre qué significa realmente este término en diseño o desempeño clínico. La propuesta intenta romper ese bloqueo con un cambio muy concreto: la equivalencia biológica puede basarse en “los mismos o similares materiales o sustancias”; y, la equivalencia clínica, puede justificarse para “la misma o similar condición clínica o propósito”.
Este matiz, “similar”, no es un adorno lingüístico; es una redefinición de lo que se considera aceptable como fuente de evidencia. En términos de diseño y desarrollo, abre una vía realista para productos que no pretenden ser disruptivos, sino versiones mejoradas: optimizaciones de usabilidad, cambios de interfaz, ajustes de performance, ampliaciones de la población de uso previsto …. En esos casos, si el salto tecnológico no introduce un perfil de riesgo distinto, la equivalencia deja de ser un muro y vuelve a ser lo que debería: una herramienta para evitar repetir estudios cuando el conocimiento existente, bien argumentado, ya responde a la pregunta clínica.
Ahora bien, “similar” no puede convertirse en “parecido” de forma vaga. Lo que cambia es que la discusión ya no debería girar alrededor de si dos productos son idénticos (que casi nunca lo son), sino alrededor de si las diferencias son relevantes clínicamente y si están controladas desde el diseño. Y ahí es donde vuelve con fuerza la ingeniería comparativa como disciplina central.
Dispositivos de tecnología bien establecida (WET): un carril más proporcional
La propuesta introduce un concepto que, si se aplica con coherencia, puede tener un efecto muy práctico en el desarrollo: el well-established technology device (WET). La idea de fondo es sencilla: no tiene sentido exigir el mismo esfuerzo de verificación/validación a productos basados en tecnologías maduras y a dispositivos innovadores. El texto define WET con criterios concretos, que en esencia apuntan a cuatro atributos: un diseño simple o estable, ausencia de historial relevante de problemas de seguridad, un rendimiento (performance) bien conocido, y una larga historia de uso en el mercado de la UE.
En clave de desarrollo, WET no es una “etiqueta bonita”: es un marco de posicionamiento del producto. Si un fabricante puede demostrar que su producto es WET, la discusión con un organismo notificado se desplaza desde “demuéstrame de nuevo lo que ya se sabe” hacia “demuéstrame que tu producto encaja de verdad en el estado del arte de una tecnología y que no has introducido novedad relevante sin control”. Esto, en la práctica, puede traducirse en exigencias más proporcionales en evaluación clínica/investigación, siempre que el fabricante haga los deberes de forma impecable.
En productos con componente digital, WET es posible, pero resulta más complicado. El mercado y la seguridad empujan a actualizaciones frecuentes (ciberseguridad, interoperabilidad, correcciones). Si el producto incorpora IA, el argumento WET se debilita salvo que el fabricante implemente un gobierno del ciclo de vida extremadamente sólido basado en criterios preestablecidos para gobernar cambios, una monitorización poscomercialización diseñada como parte del producto, y una narrativa regulatoria consistente sobre por qué los cambios no rompen la condición de “tecnología bien establecida”.
Evidencia clínica/performance: menos “papeles”, misma exigencia estructural
En mi opinión, este es uno de los puntos donde más fácil es interpretar mal el paquete. La tentación es leer “simplificación” como “rebaja” y concluir que se reduce la evidencia clínica. Pero en la práctica, lo que se modifica no es la responsabilidad de demostrar seguridad y desempeño, sino la forma de construir, realizar, y presentar esa demostración ya que permite articular un paquete de evidencia más flexible basado en literatura contrastada, equivalencia/comparabilidad, datos preclínicos, evidencia del mundo real, PMS/PMCF) sin que el expediente se convierta en un laberinto de documentos redundantes. Esto es especialmente relevante en productos complejos que incorporan IA, donde el desempeño no es solo un valor estático medido “antes” del mercado, sino un comportamiento que debe sostenerse con control de cambios y vigilancia continua.
Un puente explícito entre MDR/IVDR y AI Act
Uno de los “pecados originales” de la regulación de IA en entorno sanitario ha sido el doble cumplimiento: por un lado, el fabricante estructura su sistema con lógica MDR/IVDR, QMS, gestión de riesgos, evaluación clínica/performance, PMS/PMCF, vigilancia y control de cambios,; por otro, el AI Act introduce un segundo bloque de obligaciones que, en muchos puntos, parecen hablar del mismo problema con otro vocabulario: gestión de riesgos del sistema, gobernanza de datos, trazabilidad, supervisión humana, monitorización y controles a lo largo del ciclo de vida. El resultado típico ha sido ineficiente: documentación duplicada, responsables internos “en paralelo” y, lo peor, incoherencias involuntarias (lo que en MDR se justifica de una forma, en AI Act se expresa de otra).
Aquí conviene ser muy claro: para productos con IA, no hay barra libre. Lo que está haciendo la propuesta no es “relajar” la Ley de IA, sino encajarla dentro del ecosistema sanitario para que no funcione como una segunda capa desconectada. La propuesta intenta corregir esto con un “enganche” operativo que es más importante de lo que parece: establece que, cuando la Comisión adopte actos de ejecución o delegados o Especificaciones Comunes que afecten a dispositivos que sean sistemas de IA de alto riesgo, o que usen IA de alto riesgo como componente de seguridad, deberá tener en cuenta los requisitos del AI Act (Capítulo III, Sección 2). En términos prácticos, esto es una señal regulatoria: no se pretende crear dos mundos, sino forzar una alineación estructural para que, cuando se concreten requisitos técnicos (por ejemplo, qué se considera suficiente para monitorización, control de cambios, validación, robustez, trazabilidad), no nazcan en contradicción con lo que ya exige el AI Act.
La estrategia sensata pasa por construir un único sistema documental y operativo que soporte ambos marcos:
- MDR/IVDR: ISO 13485, ISO 14971, GSPR, V&V, usabilidad, PMS/PMCF, vigilancia, CAPA, control de cambios.
- AI Act: gobernanza de datos (calidad, representatividad, sesgos), trazabilidad del modelo, gestión del riesgo del sistema, supervisión humana, monitorización post-deployment, ciberrobustez, y disciplina de cambios (incluyendo reentrenamientos, recalibraciones, actualizaciones).
Tramitación
Dado que la propuesta introduce modificaciones en dos Reglamentos vigentes, su base jurídica se fundamenta en los artículos 114 y 168, apartado 4, letra c), del Tratado de Funcionamiento de la Unión Europea (TFUE). Por un lado, el artículo 114 del TFUE permite a la Unión Europea armonizar la legislación para garantizar el buen funcionamiento del mercado interior. En el ámbito de los productos sanitarios y de diagnóstico in vitro, esta base jurídica es esencial para evitar diferencias regulatorias entre Estados miembros que fragmentan el mercado, generan inseguridad jurídica y dificultan el acceso de los productos a los mercados de los distintos países de la Unión. Por otro lado, el artículo 168 faculta a las instituciones europeas para adoptar normas comunes de calidad y seguridad en materia de productos sanitarios, con el objetivo de garantizar un alto nivel de protección de la salud humana.
Para que el MDR/IVDR 2.0 se adopte, la propuesta de la Dirección General de Salud y Seguridad Alimentaria de la Comisión debe superar el llamado ‘’procedimiento legislativo ordinario’’. En virtud de este, la Comisión Europea, institución que ostenta el derecho de iniciativa legislativa, presenta su propuesta al Consejo de la UE y al Parlamento Europeo. Así, el Parlamento Europeo examina la propuesta, pudiendo aprobarla o presentar enmiendas. Tras ello, el Consejo de la UE tiene dos opciones: aceptar la posición del Parlamento o modificar la postura de este. En el primer caso, la propuesta sería adoptada, y en el segundo, se llevaría a cabo una segunda lectura por parte del Parlamento Europeo. En este sentido, la primera lectura no tiene una limitación temporal, pudiendo extenderse en el tiempo más de lo deseado.
La segunda lectura, sin embargo, sí tiene una duración limitada, pudiendo llegar a tomar hasta 8 meses. En este escenario, el Parlamento puede aprobar la propuesta, proponer enmiendas o rechazarla definitivamente. Si el Parlamento la aprueba o enmienda, llegado el turno del Consejo podrá aprobarlo también a su vez o no aceptar la enmienda, entrando en juego un cuarto actor: el Comité de Conciliación.
Este Comité, compuesto por un número igual de representantes del Parlamento y del Consejo deberá votar para la aprobación de la propuesta en el plazo de 6 semanas. En el caso del Parlamento, por mayoría absoluta, y en el caso del Consejo, por mayoría cualificada. De no alcanzar estas mayorías, no se adoptaría la propuesta. Si se consiguiera llegar a dichas mayorías, se procedería a una tercera y última lectura de 6 semanas de duración.
Durante el proceso, también puede darse el conocido ‘’diálogo tripartito’’, por el cual el Consejo, el Parlamento y la Comisión se reúnen de manera informal con el fin de llegar a un acuerdo que deberá aprobarse con arreglo a las normas respectivas del Parlamento y el Consejo para que sea de naturaleza vinculante.
Tras la aprobación formal y su publicación en el Diario Oficial, llega la fase decisiva: entrada en vigor y aplicación, que depende de actos delegados/de ejecución, guías (MDCG) y, sobre todo, de cómo lo implementen organismos notificados y autoridades. Por eso, los fabricantes deben seguir diseñando con el marco vigente, pero preparando el expediente para aprovechar el nuevo mecanismo de evaluación de la equivalencia, WET y la posibilidad de realizar sandboxes regulatorios sin rehacerlo. De momento, los ciudadanos y las partes interesadas tienen la oportunidad de realizar comentarios sobre la propuesta al menos hasta mediados de marzo.
Cierre: una mejora real, con letra pequeña
MDR 2.0 e IVDR 2.0 apuntan en la dirección correcta: reconocen que el problema ya no es solo “cumplir”, sino poder hacerlo con plazos, costes compatibles con la innovación. Las dos palancas más prometedoras, equivalencia “igual o similar” y el concepto de tecnología bien establecida, pueden devolver eficiencia al desarrollo, siempre que se usen con rigor y que los organismos notificados las apliquen de forma consistente.
La condición para que funcione es más operativa: claridad, coherencia y aplicación consistente. Si se consigue, el efecto no será solo regulatorio. Porque, en última instancia, las reglas no solo evalúan productos; también los moldean.
Descarga sugerida:
Artículo escrito por:
Miguel Ángel Campanero Martínez y María Sanchez Besga
Director Técnico y Técnico Jurídico Senior de Gobernanza, respectivamente
A3Z advanced y proyecto ''HealthData@MAD-R&I'' (Fundación para la Investigación e Innovación Biosanitaria de Atención Primaria), respectivamente