Las Buenas Prácticas Clínicas (BPC), conocidas internacionalmente como Good Clinical Practice (GCP) y publicadas por la ICH (Consejo Internacional de Armonización), son un estándar internacional de calidad ética y científica para el diseño, realización, registro y comunicación de ensayos clínicos en los que participan seres humanos.
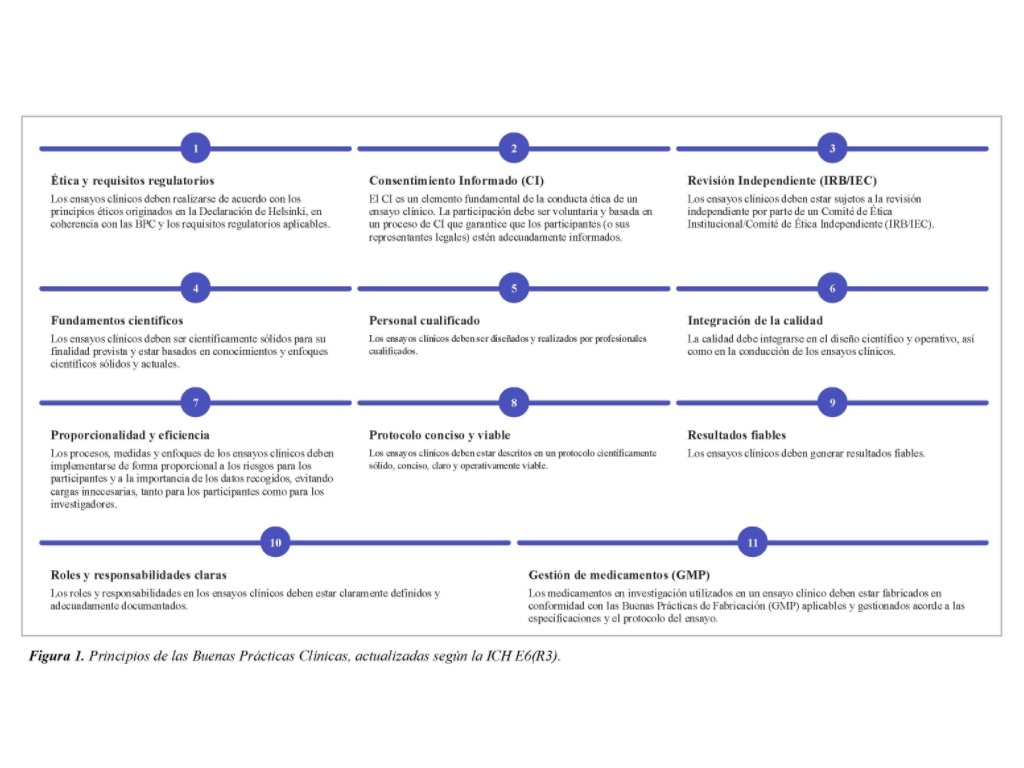
El cumplimiento de sus principios básicos (Figura 1) garantiza que los derechos, seguridad y bienestar de los participantes estén protegidos; respalda la fiabilidad y precisión de los resultados de la investigación para la toma de decisiones de salud; y asegura que la realización de los ensayos sea coherente con los principios de la Declaración de Helsinki.
Su revisión previa E6(R2) publicada en 2016, abordaba los retos asociados al avance tecnológico en la gestión de datos y reforzaba la necesidad de políticas promotoras de la transparencia y seguridad de la información, en consonancia con la globalización de los estudios clínicos y su realización en diferentes países y regiones. Esta última actualización E6(R3), diseñada para adaptarse a la creciente complejidad de los ensayos clínicos y a los avances tecnológicos, fue publicada en 2025 y representa una modernización significativa respecto a la E6(R2).
Las principales novedades recogidas en la E6(R3) afectan a diferentes aspectos de los ensayos. En primer lugar, destaca la Calidad desde el Diseño (Quality by Design, QbD), basada en un enfoque proactivo y en la identificación de factores críticos para la calidad (Critical to Quality, CtQ), fundamentales para la protección de los participantes y la fiabilidad de los resultados. En segundo lugar, la proporcionalidad y el enfoque basado en riesgos. Se establece que los procesos y estrategias de mitigación deben ser proporcionales a los riesgos para la seguridad de los participantes y la importancia de los datos recopilados. Asimismo, se insta a los promotores a reducir complejidad innecesaria en los protocolos y procedimientos para evitar cargas innecesarias a investigadores y participantes.
En tercer lugar, se aborda la digitalización y las nuevas tecnologías, adoptando una posición neutral con respecto al tipo de herramientas utilizadas para la recogida de datos, permitiendo su uso independientemente de si son en papel o electrónicas. Se incorpora el uso de tecnologías sanitarias digitales (p. ej.: wearables) que permiten capturar datos de los participantes en su entorno habitual. Además, se mencionan explícitamente por primera vez los ensayos descentralizados como una opción de diseño válida.
En cuarto lugar, se crea una nueva sección dedicada a la gobernanza de datos, que cubre todo su ciclo de vida: desde su captura hasta su destrucción. Se da una importancia crítica a los metadatos y a la interpretación de los registros de auditoría, garantizando la trazabilidad de los cambios realizados. Además, se establecen requisitos detallados para la validación y seguridad de los sistemas informatizados.
En quinto lugar, se incluyen novedades sobre la participación de las partes interesadas. Se recomienda obtener la perspectiva de los pacientes durante el diseño del ensayo para mejorar su viabilidad y la relevancia de los resultados. Además, se enfatiza la comunicación de los resultados a los participantes de forma objetiva y no promocional. Por último, en relación con la claridad en las funciones y responsabilidades, se refuerza el marco de supervisión (oversight) cuando se delegan actividades en proveedores de servicios, enfatizando que la responsabilidad última pertenece al promotor o al investigador, según corresponda. Asimismo, se permite el uso de métodos remotos y soportes multimedia para el procedimiento de consentimiento informado.
El impacto de la E6(R3) sobre la figura del promotor de los ensayos clínicos radica en la necesidad de implementar nuevas estrategias de calidad y monitorización, así como la planificación de la supervisión de sus proveedores de servicios. En paralelo, la E6(R3) afecta también a los comités de ética, los cuales deben adoptar un enfoque basado en el riesgo y la protección del paciente en sus evaluaciones. Asimismo, se les requiere prestar atención a la documentación crítica y deben evaluar protocolos adaptados al uso de tecnología y datos electrónicos.
Sin duda, estos cambios suponen un reto, destacando la necesidad de adaptación de los Planes Normalizados de Trabajo (PNT), la formación específica y de calidad al personal implicado, o la utilización de sistemas electrónicos seguros. Sin embargo, también suponen una oportunidad para aumentar la eficiencia en la ejecución de los ensayos, obtener datos confiables y mejorar la experiencia de los pacientes.
En definitiva, la E6(R3) supone una modernización transformadora de la investigación clínica, diseñada para integrar los ensayos clínicos de manera más fluida en la práctica clínica actual y en el entorno digital, manteniendo siempre el cumplimiento fundamental de los principios éticos la Declaración de Helsinki.
Bibliografía:
- International Council for Harmonisation (ICH). ICH E6 (R3) Guideline for good clinical practice (GCP) Step 5. Amsterdam: European Medicines Agency; 2025.
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Normas de Buena Práctica Clínica ICH E6 (R3). Madrid: AEMPS; 2025
Descarga sugerida:
Artículo escrito por:
Marta Valverde
Clinical Affairs
Konexio Biotech