La Asociación A3P (a3p.org) es una entidad clave de la industria farmacéutica y biotecnológica. Desde 1986, contribuye a mejorar el conocimiento de los procesos que intervienen en la fabricación de medicamentos y en el desarrollo de equipos e intalaciones en entornos asépticos, a través de su red de proveedores y fabricantes industriales, representados a partes iguales en su Consejo de Administración. Actualmente cuenta con miles de miembros que comparten su experiencia técnica, científica y regulatoria a través de eventos formativos y de networking, de CIG (Grupos de Interés Común), de la página web y de la revista La Vague, publicada trimestralmente. A3P tiene presencia internacional en filiales que son dirigidas por representantes locales. La red se impulsa mediante eventos (foros, jornadas técnicas, conferencias, talleres, exposiciones, visitas a centros farmacéuticos, etc.) organizados para responder a los temas que localmente son de más actualidad.
A continuación, A3P España, ha traducido un artículo publicado originalmente en su revista LA VAGUE.
Con la entrada en vigor de la última versión del Anexo 1, siguen existiendo dudas sobre su aplicabilidad a la producción de medicamentos de terapia avanzada (MTA). Entonces, ¿cómo seguir trabajando en este periodo de transición, a la espera de la revisión de la parte IV en 2026?
La gran diversidad de procesos, la rápida evolución de las tecnologías y el tamaño a veces reducido de los lotes de producción son las principales razones de la existencia de guías normativas específicas para la producción de medicamentos de terapia avanzada. La parte IV de las normas de correcta fabricación, que data de noviembre de 2017, es un texto considerado "autónomo" y que se basta por sí mismo.
Sin embargo, la revisión del Anexo 1, que entró en vigor el 25 de agosto del 2023, puede plantear dudas. Su ámbito de aplicación no menciona específicamente los ATMPs, pero, sin embargo, estos son, en su gran mayoría, medicamentos inyectables estériles. La aplicación de algunos artículos de este anexo es complicada, por no decir imposible, en determinados procesos/productos. El anexo 2A del PIC/S, publicado en abril de 2021, había identificado los vínculos y las excepciones del anexo 1.
A la espera de la revisión oficial de la Parte IV, hemos aplicado un enfoque similar para intentar aclarar los elementos aplicables y los que lo son menos. Como era de esperar, varios requisitos del Anexo 1 ya estaban incluidos en la parte IV. Su aplicación se detalla y explica en dicha parte. El enfoque de análisis de riesgos es fundamental, tanto para el Anexo 1 como para la parte IV. Debe abarcar todos los temas y procesos, y está estrechamente relacionado con el sistema de calidad farmacéutica. Los requisitos en materia de locales, equipos y vigilancia medioambiental también presentan numerosas similitudes, aunque el Anexo 1 es mucho más preciso. Otros temas pueden resultar más confusos: uso de RABS y aisladores, frecuencia de las APS, control de integridad al 100 %, inspección visual, etc. Dependiendo del proceso de fabricación de los ATMPs, la aplicación puede resultar insuperable. No obstante, deben aplicarse dos principios fundamentales:
- Cuanto más se acerque el proceso de fabricación de los ATMPs a un proceso estándar de bioterapia, más importante será la aplicación del Anexo 1.
- Se deberá identificar cualquier desviación y los métodos de control de riesgos deberán documentarse y justificarse mediante un análisis de riesgos.
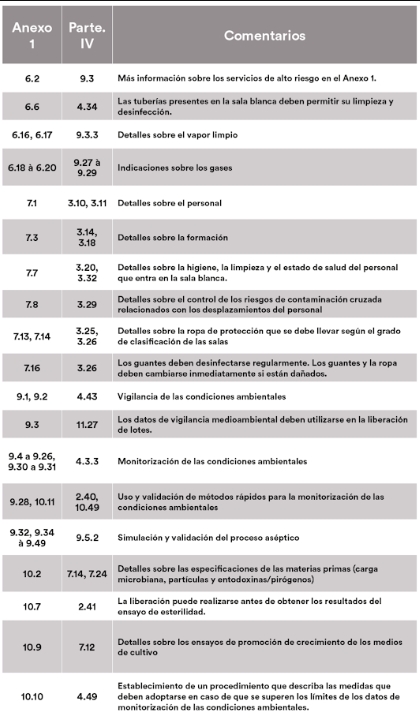
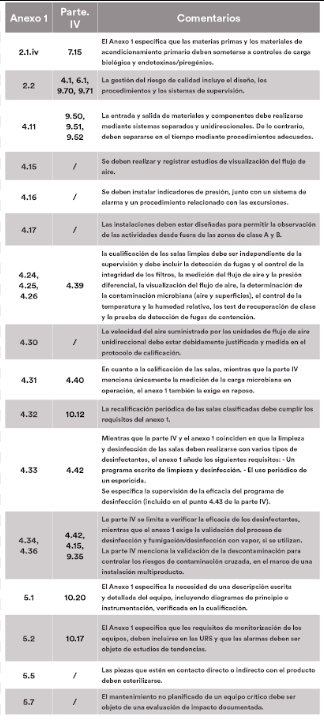
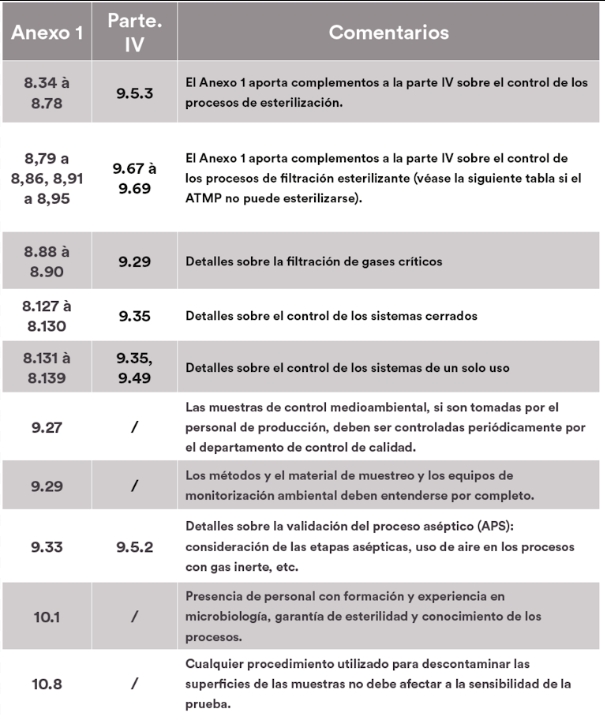
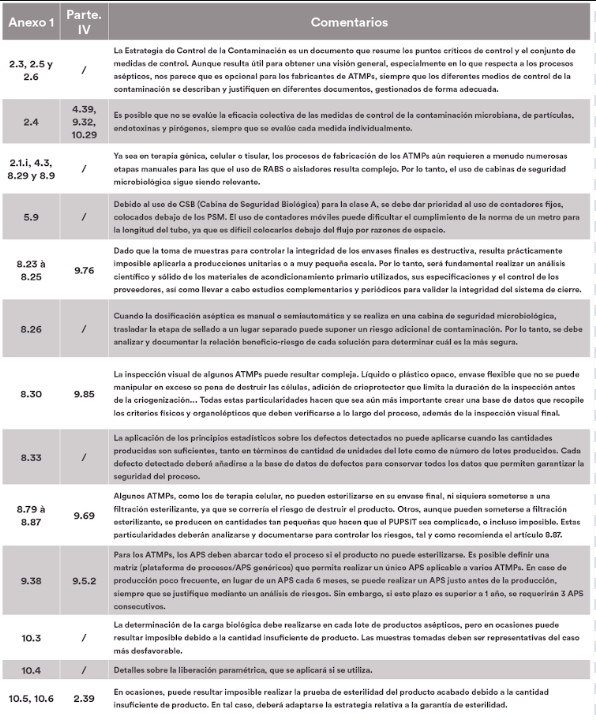
El artículo se divide en tres partes. La primera enumera los puntos cubiertos por el anexo 1 y la parte IV, la segunda detalla los puntos del anexo 1 que serían aplicables a las ATMPs, mientras que la última expone los puntos que podrían considerarse opcionales. Para facilitar la lectura y las referencias cruzadas, los datos se presentan en forma de tablas, en las que se muestra la correspondencia entre los números de los artículos del anexo 1 y de la parte IV, acompañados de nuestros comentarios.
Puntos en común
Ambos textos tienen por objeto garantizar la calidad, la seguridad y la eficacia de los medicamentos mediante el establecimiento de normas de fabricación rigurosas. Hacen hincapié en la importancia de los sistemas de gestión de la calidad, incluida la validación de los procesos, el control de calidad y la garantía de calidad. Se destacan los estrictos requisitos de higiene para evitar cualquier contaminación, tanto del personal como de los productos.
Los puntos aplicables del Anexo 1
El anexo 1 hace hincapié en la importancia de adoptar un enfoque sistemá-tico para identificar y evaluar los riesgos potenciales a lo largo del ciclo de vida del producto, así como en la necesidad de garantizar que los datos generados a lo largo del proceso de fabricación sean precisos, completos y fiables. Estos elementos, aunque implícitos en la parte IV, requieren una atención especial para garantizar la conformidad de los ATMPs. En general, se deben seguir las recomendaciones relativas a tecnologías/técnicas específicas cuando se utilicen (puntos 4.18 a 4.22, capítulo 8).
De lo contrario, la implementación de un análisis de riesgos debe permitir definir y justificar la eficacia de los medios alternativos utilizados para controlar los riesgos.
Los puntos opcionales
Algunos requisitos relativos a los equipos y las infraestructuras pueden no ser pertinentes para los medicamentos de terapia avanzada, que, en algunos casos, requieren instalaciones específicas adaptadas a procesos y métodos de fabricación únicos. Del mismo modo, los requisitos estandarizados para la fabricación de productos comerciales pueden no ser directamente aplicables a los ATMPs, que requieren una mayor flexibilidad en los métodos y protocolos de producción. Los procedimientos tradicionales de control y garantía de calidad pueden adaptarse a las terapias avanzadas debido a la naturaleza evolutiva y experimental de estos productos.
En conclusión
Muchos de los artículos del Anexo 1 son aplicables y útiles para la fabricación de ATMPs estériles. Sin embargo, la especificidad de los procesos, las tecnologías y los productos hace que, en ocasiones, su aplicación sea imposible. Estas diferencias reflejan la naturaleza única de la producción de terapias innovadoras, que a menudo requiere métodos personalizados y enfoques flexibles en comparación con los estándares de fabricación tradicionales. Es fundamental que las autoridades reguladoras y los fabricantes adapten las buenas prácticas en consecuencia, manteniendo al mismo tiempo la seguridad y la eficacia de los productos.
La EMA ha anunciado que el grupo de trabajo de inspectores se reunirá con el CAT (Comité de Terapias Avanzadas) y la Comisión Europea a partir del último trimestre de 2026 para estudiar la necesidad de revisar las directrices a la luz del nuevo Anexo 1: Fabricación de medicamentos estériles. Mientras tanto, el uso de herramientas de análisis de riesgos y el establecimiento y registro de datos científicos deben permitir demostrar la solidez de los procesos utilizados para preservar la asepsia.
Descarga sugerida:
Artículo escrito por:
Gaëlle Caplat y Sophie Michel
CellforCure by SEQENS y SMichel Consulting, respectivamente
A3P