Los medicamentos de terapia avanzada (ATMPs, por sus siglas en inglés: Advanced Therapy Medicinal Products) son tratamientos para humanos que están basados en genes, células o tejidos [1]. Los ATMPs representan una de las innovaciones más significativas en la medicina regenerativa contemporánea, ya que están transformando de forma radical el abordaje de un amplio rango de enfermedades difíciles de curar de manera convencional.
A diferencia de los tratamientos farmacológicos tradicionales que se centran principalmente en el alivio de los síntomas, estas terapias abordan la causa subyacente de la enfermedad aprovechando mecanismos biológicos para reparar, reemplazar o regenerar tejidos, órganos o células dañadas. Se trata de una revolución en el área de la medicina y de la manufactura farmacéutica, que redefine las posibilidades actuales terapéuticas.
Bajo el término ATMP se agrupan cuatro tipos de productos terapéuticos diferenciados. Por un lado, tenemos la terapia génica, que implica la inserción, modificación o eliminación de material genético con el objetivo de tratar o prevenir enfermedades. Por otro lado, la terapia celular, que emplea células humanas que han sido sometidas a una manipulación sustancial modificando sus propiedades biológicas, fisiológicas o estructurales con fines terapéuticos, diagnósticos o preventivos. En tercer lugar, se encuentra la ingeniería de tejidos, en la que se emplean células y/o tejidos manipulados para regenerar, restaurar o sustituir estructuras biológicas dañadas, cuando están destinados a desempeñar las mismas funciones que en el donante. Finalmente, los ATMPs combinados integran uno o más productos sanitarios (dispositivos médicos), con algún tipo de actividad o no, junto con células o tejidos (viables o no), que actúan de manera sinérgica para cumplir una función terapéutica [1].
A pesar de su enorme potencial, el desarrollo, autorización y comercialización de estos medicamentos plantea retos regulatorios y científicos significativos. Es un campo en rápida expansión, muy heterogéneo en cuanto a tipo de producto medicinal y enfermedad a tratar y con mucha complejidad técnica, clínica y normativa. Por ello, resulta esencial comprender cómo se estructura su regulación y qué mecanismos existen para garantizar su seguridad, eficacia y calidad, los tres pilares fundamentales sobre los que se asienta la evaluación de cualquier medicamento de uso humano en Europa.
En este contexto, la regulación de los ATMPs en la Unión Europea ha evolucionado para ofrecer vías específicas de acceso al mercado, tanto a través de procedimientos estándar como de mecanismos excepcionales. Este artículo ofrece una visión de los principales modelos de autorización existentes en Europa, incluyendo la autorización de comercialización centralizada, el uso compasivo, la exención hospitalaria, así como las diferencias entre los procedimientos de autorización aplicables a estos productos. Comprender este entramado normativo es clave para facilitar el desarrollo, acceso y disponibilidad para este tipo de terapias que pueden representan la única opción para los pacientes.
Autorización de comercialización: La vía centralizada para ATMPs en la UE
La autorización de comercialización (Marketing Authorisation, MA) constituye el proceso estándar indispensable para que un ATMP pueda ser comercializado en la Unión Europea. Este trámite se realiza exclusivamente mediante el procedimiento centralizado, gestionado por la Agencia Europea de Medicamentos (EMA). Una vez obtenida la autorización, se permite el acceso al producto para los 27 Estados miembros de la UE, Islandia, Liechtenstein y Noruega (que forman parte del Espacio Económico Europeo, EEE), garantizando un acceso unificado al mercado europeo con una sola solicitud [2].
El objetivo primordial de la fase de desarrollo clínico del ATMP es precisamente obtener esta autorización. Durante este período, el solicitante recopila y presenta evidencias que demuestran la eficacia, seguridad y calidad del ATMP, integradas en un expediente técnico riguroso. La EMA evalúa estos datos para emitir una decisión basada en el balance beneficio-riesgo, requisito fundamental consagrado en la legislación europea [3,4].
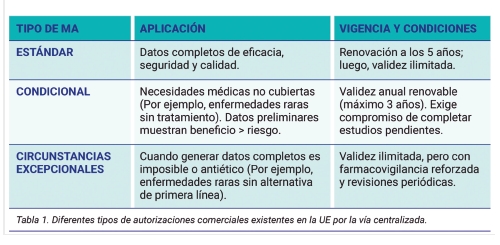
La complejidad inherente a los ATMPs ha impulsado a la EMA a desarrollar un sistema de autorización multifacético. Este modelo reconoce que la generación de evidencia completa en cuanto a eficacia y seguridad, para productos altamente innovadores enfrenta desafíos únicos, especialmente en enfermedades raras o de alta mortalidad. Así, el marco regulatorio establece más categorías para su autorización, cada una adaptada a distintos niveles de solidez científica y tipo de enfermedad [5].
La Autorización Estándar representa el nivel máximo de exigencia, aplicable cuando el desarrollador presenta datos preclínicos y clínicos concluyentes que demuestran de manera inequívoca el perfil beneficio-riesgo del producto. Este tipo de autorización, que tras una renovación a los cinco años adquiere validez ilimitada, es característico de ATMPs con ensayos clínicos robustos [6].
Frente a necesidades médicas no cubiertas donde la rapidez en el acceso puede ser determinante, la EMA dispone de la Autorización Condicional [7,8]. Este mecanismo acepta datos preliminares que sugieran un balance beneficio-riesgo positivo, siempre que el solicitante se comprometa a completar estudios post comercialización. La validez, inicialmente anual, permite que tratamientos pioneros como las terapias CAR-T para leucemias refractarias y linfomas alcancen a pacientes años antes que bajo la Autorización Estándar [9]. La transformación a Autorización Estándar ocurre únicamente cuando se verifican los resultados prometidos, garantizando así que la flexibilidad no comprometa la seguridad [10].
En el extremo de la regulación se encuentra la Autorización en Circunstancias Excepcionales, reservada para situaciones donde la recopilación de evidencia completa resulta científicamente inviable o éticamente cuestionable. Este escenario es frecuente en enfermedades muy raras con cohortes mínimas de pacientes, donde los ensayos controlados tradicionales son imposibles. La autorización, que mantiene validez ilimitada, pero bajo supervisión reforzada, exige una justificación metodológica rigurosa y un plan de farmacovigilancia activa [11,12].
Es fundamental destacar que estas categorías (Tabla 1) no comprometen nunca la calidad del producto ATMP, sino que corresponden a adaptaciones pragmáticas a realidades científicas diversas [13,14].
Proceso de autorización descentralizado en la UE
Además del procedimiento centralizado, existen procedimientos descentralizados y nacionales que permiten la autorización de medicamentos en uno o más estados miembros (Tabla 2). El procedimiento descentralizado sirve para autorizar medicamentos en más de un estado miembro de la Unión Europea en paralelo. Puede ser utilizado para medicamentos que no necesitan ser autorizados a través del procedimiento centralizado y que no han sido autorizados previamente en ningún estado miembro [22].
Accesos excepcionales descentralizados: Puentes terapéuticos para situaciones críticas
Cuando los plazos de desarrollo chocan con la urgencia clínica, la UE ha establecido vías excepcionales de acceso. El Uso Compasivo (Compassionate Use) permite administrar ATMPs no autorizados a pacientes con enfermedades graves o raras sin alternativas disponibles, siempre que no puedan participar en ensayos clínicos [23,24]. Este mecanismo, coordinado e implementado a nivel nacional bajo directrices europeas, presenta variaciones significativas entre Estados miembros. Aunque no todos los estados miembros tienen legislación y procesos bien definidos respecto el uso compasivo, la mayoría de los estados sí lo tienen [25]. Los 3 países europeos que mejor representan este tipo de autorización en cuanto a la regulación europea y nacional son: Alemania, Italia y España, además del Reino Unido, cuyos programas de Uso Compasivo están supervisados por sus respectivas agencias nacionales del medicamento. En Reino Unido opera el “Early Access to Medicines Scheme”, que requiere datos de fase III avanzados; en Alemania lo regula su Ley de Medicamentos (“German Medicinal Product Act“), exigiendo supervisión hospitalaria estricta; mientras Italia lo contempla en su “Decreto Ministeriale” de 2017 para enfermedades graves o raras. En España lo regula el Real Decreto 1015 del 2009 [26] y el acceso está supervisado por la Agencia Española del Medicamento y productos sanitarios (AEMPS). La EMA, aunque no administra los programas, emite opiniones científicas sobre protocolos de uso para garantizar coherencia en seguridad entre países [25].
Complementariamente, la Exención Hospitalaria (Hospital Exemption) [27], autoriza la fabricación y uso de ATMPs sin autorización comercial en entornos hospitalarios específicos. Esta figura, crucial para terapias autólogas o personalizadas, se rige por cuatro pilares:
- Debe tratarse de preparados no rutinarios fabricados bajo normas GMP: excluye producción industrial o procesos estandarizados. Sin embargo, la falta de definición UE de "no rutinario" ha generado interpretaciones nacionales divergentes.
- Administrados exclusivamente dentro de una misma institución: restringe la administración al hospital fabricante, limitando la escalabilidad y generando inequidades geográficas.
- Bajo responsabilidad médica directa: el médico prescribe y supervisa el tratamiento de forma individualizada.
- Destinados a pacientes individuales: la ambigüedad en "custom-made" lleva a disparidades en su aplicación. Por ejemplo, mientras España requiere prescripciones nominativas, Finlandia acepta pequeños lotes para patologías similares.
.jpg)
En España, la AEMPS exige evaluación de datos preclínicos, acreditación de instalaciones, y sistemas de trazabilidad equivalentes a los requeridos para productos comerciales [28].
No obstante, la vía de Exención Hospitalaria enfrenta retos sustanciales de fragmentación regulatoria. Los criterios para implementar la exención varían significativamente entre estados miembros, generando inequidades en el acceso. Mientras Francia limita su aplicación a enfermedades raras sin tratamiento autorizado, otros países permiten usos más amplios, como por ejemplo en Italia, donde no hay límite numérico de pacientes [29]. Esta disparidad ha impulsado iniciativas de armonización en el nuevo código farmacéutico europeo (en trámite), que estandarizarán requisitos de calidad y supervisión para la Exención Hospitalaria sin comprometer su flexibilidad. La propuesta incluye tener un registro centralizado en la UE para ATMPs bajo exención hospitalaria, criterios unificados para la definición de “preparados no rutinarios” y reportes obligatorios de seguridad. Estas medidas, derivadas del Reglamento (CE) 1394/2007 [30], abordarán la fragmentación actual manteniendo la flexibilidad necesaria para mantener acceso a terapias personalizadas, estandarizando requisitos de calidad y supervisión.
Conclusión: Equilibrio dinámico entre innovación y garantías
El ecosistema regulatorio europeo para ATMPs refleja una evolución constante hacia modelos que reconcilian la urgencia médica con el rigor científico. Las vías flexibles de autorización, los programas de apoyo al desarrollo y los mecanismos de acceso temprano constituyen respuestas adaptativas a las singularidades de estas terapias transformadoras. España, mediante la AEMPS, ha implementado eficientemente estos marcos, aunque persisten desafíos en la armonización de criterios nacionales, especialmente en exención hospitalaria y uso compasivo. El futuro próximo probablemente testificará mayor convergencia internacional, impulsada por el crecimiento exponencial de plataformas tecnológicas como la edición génica o las células iPSC, que demandarán marcos ágiles sin comprometer los estándares de seguridad que caracterizan al modelo europeo. La colaboración continua entre reguladores, desarrolladores y clínicos seguirá siendo esencial para convertir el potencial científico en realidades terapéuticas accesibles.
.jpg)
Bibliografía
- [1] https://www.ema.europa.eu/en/human-regulatory-overview/advanced-therapy-medicinal-products-overview.
- [2] https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation/advanced-therapies-marketing-authorisation.
- [3] Directiva 2001/83/CE.
- [4] Reglamento (CE) 726/2004.
- [5] https://www.ema.europa.eu/en/human-regulatory-overview/advanced-therapy-medicinal-products-overview/legal-framework-advanced-therapies.
- [6] https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation/advanced-therapies-marketing-authorisation.
- [7] Reglamento (CE) 507/2006.
- [8] Reglamento (CE) 726/2004.
- [9] https://www.ema.europa.eu/en/news/first-car-t-cell-medicine-mantle-cell-lymphoma#:~:text=EMA%20has%20recommended%20granting%20a%20conditional%20marketing%20authorisation,after%20two%20or%20more%20lines%20of%20systemic%20therapy.
- [10] https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation/conditional-marketing-authorisation#:~:text=EMA%27s%20CHMP%20may%20grant%20a%20conditional%20marketing%20authorisation,the%20fact%20that%20additional%20data%20are%20still%20required.
- [11] Reglamento (EC) 726/2004.
- [12] https://www.ema.europa.eu/en/human-regulatory-overview/marketing-authorisation/pre-authorisation-guidance#1-types-of-applications-and-applicants-6893.
- [13] Reglamento (CE) 507/2006.
- [14] RD 1345/2007.
- [15] https://www.ema.europa.eu/en/glossary-terms/decentralised-procedure
- [16] Regulation (EC) 726/2004
- [17] https://www.ema.europa.eu/en/human-regulatory-overview/research-development/compassionate-use
- [18] Balasubramanian G, Morampudi S, et al. An overview of Compassionate Use Programs in the European Union member states; Intractable Rare Dis Res.; 2016 Nov;5(4):244-254.
- [19] RD 1015/2009
- [20] Reglamento (CE) 1394/2007
- [21] RD 477/2014
- [22] Hills, Allison et al.; An assessment of the hospital exemption landscape across European Member States: regulatory frameworks, use and impact; Cytotherapy; Volume 22, Issue 12, 772 – 779.
- [23] Reglamento (CE) 1394/2007
Descarga sugerida:
Artículo escrito por:
Núria Nieto, Jordi Gibert y Andrea Romero
Advanced Therapy Lead, Head of Biotechnology Business Unit, y Quality Assurance & Regulatory affairs in ATMPs, respectivamente
Klinea Biotech & Pharma Engineering y Konexio Biotech.